Online CE Programs
A Roundtable Discussion on Complement 3 Glomerulopathy (C3G): Improving Diagnosis, Risk Stratification, and Therapeutic Strategies
Complement 3 glomerulopathy (C3G) is a rare, progressive kidney disease characterized by dysregulation of the alternative pathway (AP) of the complement cascade, either due to genetic variants or acquired humoral factors1. About 50% of patients with C3G progress to kidney failure within 10 years of diagnosis2,6. Diagnosis and management are frequently delayed in part due to overlapping clinical presentations, histologic features, and underlying pathophysiologies.
This on-demand CME webinar will review illustrative patient cases, discussing consensus guideline treatment recommendations and the uncertainties, challenges, and considerations regarding the diagnosis and management of patients with C3G in clinical practice using an interactive roundtable conversation format.

Early Recognition and Management of Dupuytren's Contracture
This CME activity will provide a practical overview of Dupuytren’s contracture, a common hand pathology that is often first present in the primary care setting. The session will focus on recognizing key clinical features, performing effective diagnosis and screening in everyday practice, and understanding available treatment options ranging from observation and non-surgical interventions to surgical management. Emphasis will be placed on the role of the osteopathic internists in early detection, patient education, and timely referral to hand specialists when appropriate.
Exploring Biosimilars in Pediatric Psoriasis: Emerging Treatment Options
Biosimilars are complex biologic therapies derived from living organisms and developed to be highly similar to an FDA-approved reference product in structure, function, and clinical performance, without clinically meaningful differences in safety, purity, or potency. This activity will review current advancements in the understanding and clinical application of biosimilars for the management of psoriasis. Growing evidence supports their safety, efficacy, and real-world integration, highlighting biosimilars as cost-effective alternatives to originator biologics and important tools for expanding patient access to high-quality biologic treatments.

Highlights from ACEP - Severe Abdominal Pain: Could it be an Acute Porphyria Attack?
Patients with certain inherited metabolic disorders often arrive in the ED with porphyria-like attacks. To better understand AHP (acute hepatic porphyria) whose main acute presentations include severe abdominal pain, extensive neuropathy issues, disabling respiratory symptoms, tachycardia, vomiting, and more, patient cases will be reviewed covering these neurovisceral acute attacks, with recommended strategies for immediate patient treatment will be discussed.

Highlights from the Calif Neurology Society - Acute Hepatic Porphyrias: Recognition, Differential Diagnosis, and Treatment Strategies
Acute hepatic porphyrias (AHP) are rare, inherited metabolic disorders characterized by life-threatening neurovisceral attacks and chronic, often misdiagnosed symptoms. These disorders frequently present with nonspecific symptoms such as severe abdominal pain, neuropathy, vomiting, respiratory distress, and tachycardia, making accurate and timely diagnosis challenging. This webinar includes highlights from a live-in person presentation held at the California Neurology Society and will provide tools to recognize AHP, distinguish it from other acute presentations with similar features, and apply evidence-based treatment strategies. Using real patient cases, participants will gain a deeper understanding of the clinical spectrum of AHP and be better prepared to confidently identify and treat AHP in all care settings.

Highlights from the EIM Annual Meeting: Acute Hepatic Porphyria in the ED – Spotting the Red Flags behind Severe Abdominal Pain
Acute hepatic porphyrias (AHPs) are rare, inherited metabolic disorders that can present severe, recurrent episodes of abdominal pain accompanied by a wide range of nonspecific gastrointestinal, neurologic, psychiatric, and autonomic symptoms. Because these manifestations often mimic more common conditions, patients frequently experience prolonged diagnostic delays, multiple healthcare encounters, and unnecessary procedures before an accurate diagnosis is established. Through a case-based discussion, faculty will review the clinical features that distinguish AHP from more common causes of abdominal pain, highlight key diagnostic clues and red flags, and discuss appropriate laboratory testing and referral pathways. This activity aims to support earlier diagnosis, reduce diagnostic delays, and improve outcomes for patients with this potentially life-threatening condition.
Hypophosphatasia (HPP): Proposed HPP International Working Group Criteria
Hypophosphatasia (HPP) is a progressive, debilitating and sometimes fatal metabolic bone disease caused by loss of function mutations in the ALPL gene. Its clinical manifestations and severity of symptoms vary widely from patient to patient.
A multinational consortium of key opinion leaders, who are world specialists in hypophosphatasia (HPP), convened virtually during the past couple of years to recommend criteria for the diagnosis of HPP in children and adults, given that there are no formal diagnostic guidelines HPP.
Our two presenters are co-authors of the Proposed Criteria and will offer a review of the criteria including the measurable factors or parameters used to determine them, consensus recommendations for clinical practice, and appropriate treatment. By providing practical information and tools to help healthcare professionals better understand the diagnostic process, patients can be diagnosed earlier and experience better disease management and quality of life.
Metachromatic Leukodystrophy (MLD): New Consensus Guidelines for Monitoring and Management
Metachromatic leukodystrophy (MLD) is a rare, inherited lysosomal storage disorder (LSD) that is fatal, and progressive caused by biallelic pathogenic mutations in the arylsulfatase A (ARSA) gene. As a neurodegenerative disorder, its clinical manifestations primarily affect central and peripheral nervous systems, resulting in progressive loss of motor and cognitive functions, eventually leading to severe disability and early death if untreated.
A multinational consortium of key opinion leaders, who are specialists in MLD, convened virtually during the past couple of years to recommend guidelines and best-practice recommendations based on healthcare resources in the US for a standardized approach to diagnosis, presymptomatic monitoring, and clinical care.
Our two presenters are co-authors of the Consensus Recommendations and will offer a review of practice recommendations for clinical management of presymptomatic and symptomatic patients. By providing practical information and tools to help healthcare professionals better understand the diagnostic process, patients can be diagnosed earlier and experience better disease management and improved clinical care.

Overcoming Challenges in the Diagnosis and Management of Alagille Syndrome
Despite growing recognition of Alagille syndrome (ALGS) as a leading cause of pediatric cholestatic liver disease, significant gaps remain in HCP knowledge and practice related to timely diagnosis, comprehensive multisystem evaluation, and early, evidence-based management. There is limited awareness of the need for early, proactive screening for cardiac, renal, vascular, and nutritional complications, which contributes to preventable morbidity.
Join us as we discuss real patient cases covering rapid advances in medical management using targeted therapies for cholestatic pruritus and evolving best practices for nutritional and growth optimization.
Progressive Familial Intrahepatic Cholestasis (PFIC): Genetics, Treatment, and Management
Progressive Familial Intrahepatic Cholestasis (PFIC) comprises a group of rare, autosomal recessive genetic disorders marked by impaired bile formation, resulting in the hepatic accumulation of bile acids. If unrecognized or left untreated, PFIC can progress to severe liver disease and ultimately liver failure. Although uncommon, the condition’s severity—combined with evolving insights into its genetic underpinnings, diagnostics, and therapeutic options—highlights the need for targeted clinician education to support earlier diagnosis, evidence-based management, and improved patient outcomes.
Join us for an interactive panel discussion featuring real-world patient cases and expert-recommended strategies designed to address persistent clinician knowledge gaps and enhance clinical practice in PFIC care.
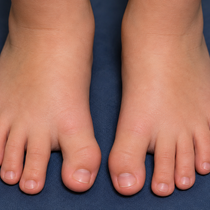
Unraveling Fibrodysplasia Ossificans Progressiva: Advances in Diagnosis, Management, and Treatment
Fibrodysplasia ossificans progressiva (FOP) is a rare, disabling genetic disorder of connective tissue characterized by congenital malformations of the great toes and progressive heterotopic endochondral ossification (HEO), the abnormal transformation of soft tissues into bone. As the most severe form of HEO in humans, FOP often leads to cumulative immobility and significant functional impairment. Frequently misdiagnosed as cancer or fibrotic disease, the disease typically presents in early childhood, often beginning in the neck and shoulders and advancing through the trunk and limbs. Over time, muscle tissue, tendons, and ligaments harden into bone tissue that develops outside the normal skeleton, leading to joint ankylosis, progressive loss of mobility, and profound physical disability.
Our expert faculty will examine the clinical challenges and consequences of a delayed diagnosis and review strategies for accurate evaluation, timely diagnosis, and evidence-based management of FOP.